
Tartalomjegyzék
Analitikai kémia (kvantitatív)
- Az előző féléves analitika folytatása, de ezúttal mennyiségi meghatározás történik.
- Az előadás anyaga nincs sehol se leírva, így érdemes bejárni, ráadásul a tanárnő tényleg azt kérdezi ami elhangzik, illetve a szigorlati tételekhez is jó vázlatot ad. Van egy repróban kapható előadásjegyzet, amelyben szinte minden benne van, ami kellhet kvantiból.
- A ZH-kban kvantitatív kérdéseken kívül kvalitatívak is szerepelnek (kb. 25% kvali).
- Szigorlaton 2 A tételt kell húzni, ezek kvantis tételek, valamint egy B tételt (kvali), ezen kívül van egy egyszerűbb számítási feladat. Általában a tanárnő vizsgáztat kvantiból, Szoboszlai tanár úr kvaliból. A tanárnő rengeteg időt ad gondolkozni, azonban semmit nem szokott segíteni. Sokszor arra kérdez rá, amit már leírt az ember a papírjára.
- Gyakorlaton első félévben, valamint második félév első felében titrimetriás meghatározások vannak, majd az utolsó néhány hétben egy műszeres blokk következik. Gyakori az egyéni ismeretlen, amelyeket 1%-os pontossággal kell meghatározni a jeles érdemjegyhez. Ha 4% a hiba, akkor elégtelen jár. Egy félévben maximum 3 elégtelen mérés lehet, de bármelyik mérést lehet egyszer pótolni, ebben az esetben a második mérés érdemjegye számít.
- Az évfolyam ZH nem lehet elégtelen, és a ZH-k átlagának legalább 2,00-nak kell lenniük. Ennek és a mérésjegyeknek az átlaga adja a gyakorlati jegyet.
| A tárgy adatlapja | |
|---|---|
| kódja a Neptunon | GYASKAKKG1M |
| típus | előadás és gyakorlat |
| intézet | ELTE-TTK Szervetlen és Analitikai Kémiai Tanszék |
| intézet honlapja | http://webkvanti.chem.elte.hu/ |
| oktatási felelős | Csörgeiné Dr. Kurin Krisztina |
| számonkérés típusa év közben | 2 ZH + 1 évfolyam ZH |
| jegymegajánlás/tanulmányi verseny | tanulmányi versenyen helyezettek könyvutalványt kapnak (illetve a tavaszi félévben felmentést a vizsgán a számolás alól) |
| félévek száma | 2 |
| vizsga típusa | 1. félévben gyakorlati jegy, 2. félévben gyakjegy + szigorlat |
| nehézség | átlagos nehézségű tárgy |
mérések
acidi-alkalimetria
kénsav meghatározása
- a kénsav vizes oldatban kétbázisú erős savként titrálható
- NaOH mérőoldattal
- metilvörös indikátor jelenlétében
- az ekvivalenciapontban hagymaszínűből sárgába vált az oldat
- az egyenértéktömeg a molekulatömeg fele
~ 0,1N sósav mérőoldat faktorozása
- analitikai pontossággal mért KHCO3 segítségével határozzuk meg a mérőoldat pontos összetételét
- a meghatározandó sósav mérőoldattal titrálunk
- HCO3- + H+ = CO2 + H2O
- a metilvörös indikátor kezdetben sárga
- kezdődő piros színnél a titrálást megszakítva az oldat CO2-tartalmát kiforraljuk
- a titrálást folytatva már nem elhzódó, hanem éles végpontot észlelünk
- az egyenértéktömeg a molekulatömeggel egyezik meg
nátrium-hidroxid és -karbonát egymás melletti meghatározása
- kétindikátoros (Warder-féle) módszer
- az első lépésben fenolftalein indikátor mellett sósavval titráljuk a lúgot és a karbonátot együttesen tartalmazó oldatot, a fenolftalein átcsapási pH-tartományát NaCl adagolásával az alacsonyabb, kedvezőbb irányba toljuk el
- először a hidroxidion semlegesítődik a savval:
- OH- + H+ = H2O
- a karbonátion pedig hidrogén-karbonáttá alakul:
- CO32- + H+ = HCO3-
- a második titrálás során metilvörös indikátor mellett a sósav mérőoldat a hidrogén-karbonát tartalmat méri (a végpontban az oldatot kiforralva)
- HCO3- + H+ = H2CO3 (» H2O + CO2)
- az első titrálás során fogyott mérőoldat térfogatából levonjuk a második titrálásnál kapott fogyást, így kapjuk a hidroxiddal egyenértékű sav térfogatát, a második titrálásnál fogyott sav közvetlenül ekvivalens a karbonáttartalommal (a karbonátot egyértékű bázisként kezelve)
- így a NaOH és a Na2CO3 egyenértéktömege is a molekulatömeggel egyezik meg
nátrium-tioszulfát indirekt meghatározása brómos oxidáció után
- a tioszulfát brómmal (brómos vízzel) hidrogénionok képződése közben szulfáttá oxidálható:
- S2O32- + 4 Br2 + 5 H2O = 2 SO42- + 8 Br- + 10 H+
- a felszabadult savat a bróm kiforralását követően metilvörös jelenlétében NaOH mérőoldattal megtitráljuk
- a bróm teljes kiforralásáról metilvörös becseppentésével győződhetünk meg
- az egyenértéktömeg a molekulatömeg tizede
bórax meghatározása
- a bórax (tetraborát anion) sósav mérőoldattal metilvörös indikátor jelenlétében kétértékű bázisként határozható meg
- a felszabaduló bórsav igen gyenge sav, így a meghatározást jelenléte nem zavarja
- B4O5(OH)42- + 2 H+ + 3 H2O = 4 H3BO3
- színváltás sárgából vörösbe
- az egyenértéktömeg a molekulatömeg fele
- ha az ismeretlen bórsavtartalmát kívánjuk meghatározni a bóraxtartalom mellett, akkor:
- metilvörös mellett megtitráljuk sósav mérőoldattal a bóraxtartalmat
- mannitot adunk hozzá » a bórsavból keletkező komplex egyértékű, közepes erősségű sav
- fenolftalein mellett NaOH-dal titráljuk
- színváltás sárgából vörösbe, majd sárgán keresztül narancsos színig
ecetsav meghatározása potenciometriás végpontjelzéssel
- vizes oldatban csak a 10-7-nél nagyobb disszociációállandójú savak határozhatók meg
- az ecetsav Kd=1,85 * 10-5, így erős bázis mérőoldattal mérhető
- a végpontot potenciometriásan jelezzük
- a mérőelektród üvegelektród, a referenciaelektród kalomelelektród
- a titrálás során mérjük a rendszer elektromotoros erejét
- a mérőoldatfogyás függvényében ábrázoljuk az adatokat
- az egyenértéktömeg a molekulatömeggel egyezik
betainium-klorid meghatározása konduktometriás végpontjelzéssel
- a betainium-klorid:
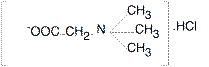
- vizes oldatban szinte tökéletesen disszociál, sósavtartalma nátronlúggal közvetlenül titrálható
- konduktometriás végpontjelzés esetén
- vezetőképességi elektródot (harangelektród) használunk
- első szakasz: a legnagyobb moláris vezetőképességű hidrogénionok koncentrációja, s ezzel együtt a mért vezetőképesség is csökken
- a mért vezetőképesség az ekvivalenciapontban a legkisebb
- az ekvivalenciapont után a hidroxidionok koncentrációjának növekedése miatt a vezetőképesség is újra nő
- a mért értékeket a fogyás függvényében ábrázoljuk
- az egyenértéktömeg a molekulatömeggel egyezik

tejsav meghatározása ("szabad" savtartalom)
- a tömény tejsavoldatokban a tejsav monomer tejsav és laktiltejsav formájában van jelen
- NaOH-dal titrálva egyértékű savként reagál a monomer tejsav:

- és a laktiltejsav is:

- NaOH mérőoldattal fenolftalein indikátor mellett halvány rózsaszínig titrálunk
- a mérés a monomer és a laktiltejsav együttes mennyiségét, a szabad savtartalmat méri
- ha ehhez a megtitrált oldathoz ismert feleslegben nátronlúgot adunk, melegítés és várakozás után (elszappanosítás) a mérőoldat feleslegét sósavval mérhetjük vissza. a két titrálásban fogyott összes NaOH mennyisége az anyag összes savtartalmának felel meg
- az egynértéktömeg a molekulatömeggel egyezik meg
lidokain nemvizes közegű meghatározása
- a lidokain
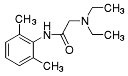
- bázisként vizes közegben nem, de jégecetes oldatban megmérhető
- a mérőoldat jégecetes perklórsav, a mintát is jégecetben kell föloldani
- kristályibolya indikátort használunk, a kékeszöld szín eléréséig
- egyenértéktömeg a molekulatömeggel egyezik meg

komplexometria, kelatometria
ólom(II)-ionok kelatometriás meghatározása
- ólomionokat pH≈10 és pH≈5 kémhatású oldatban egyaránt titrálhatunk
pH≈10
- ammónia/ammónium pufferoldattal állítjuk be a kívánt kémhatást
- a Pb(OH)2 csapadék leválását tartarátionokkal akadályozzuk meg
- eriokrómfekete-T indikátort használunk, színváltás: lilából élénk kék végleges színig
- EDTA mérőoldatot használunk

pH≈5
- a salétromsavas oldathoz urotropint adunk
- metiltimolkék indikátorral a színváltás kékből állandó sárgába
Ni(II)-ionok kelatometriás meghatározása
pH≈9-10
- ammóniával állítjuk be a pH-t
- az ammónia egyben segédkomplexképző is (annyit kell csöpögtetni, hogy a csapadék éppen föloldódjon)
- murexid indikátorral a színváltás sárgából ibolyába
- EDTA mérőoldattal
Bi(III)-ionok kelatometriás meghatározása
- a bizmutionokat a BiY- komplex nagy stabilitása miatt pH≈0,8-2,5 értékek között titrálhatjuk
- kiküszöböltnek tekinthető a bizmutionok hidrolízise és lehetőség nyílik a bizmutionok mérésére kétértékű fémionok mellett
pH≈0,8-2,5
- a kívánt kémhatást cc. salétromsavban való oldással érjük el
- metiltimolkék indikátort használunk
- EDTA mérőoldattal kékből állandó sárgáig titrálunk
- túlsavanyítás esetén az oldat liláskék, ekkor ammóniával állíthatjuk be a helyes pH-tartományt
"burow"-oldat alumíniumion-tartalmának meghatározása
- az alumínium széles pH-tartományban stabilis komplexet képez EDTA-val, a komplex kialakulása lassú
- emiatt az Al3+ közvetlenül nem, de visszaméréses titrálással meghatározható:
- ismert feleslegű EDTA jelenlétében az oldatot forraljuk, majd a mérőoldat fölöslegét visszamérjük pH≈5-nél, cinkionos mérőoldattal
pH≈5
- kénsavas oladthoz EDTA mérőoldat feleslegét adjuk és forraljuk
- hexametilén-tetramint (urotropin) adunk hozzá, továbbforraljuk
- a mérőoldatfölösleget ZnSO4-tal mérjük vissza
- metiltimolkék indikátort alkalmazva a színváltás sárgából kékig (visszamérés miatt első színváltozásig)
- ha az alumíniumot valamilyen kétértékű ion mellett akarjuk szelektíven meghatározni, a visszamérés után fluoridot adva az oldathoz az alumíniummal egyenértékű EDTA szabadul föl
kalcium és magnéziumionok egymás melletti meghatározása ásványvízben
pH≈12
- a kémhatást NaOH-dal állítjuk be
- a hidroxidionok maszkírozzák a magnéziumionokat
- a kalciumionok mérhetők murexid indikátor jelenlétében EDTA mérőoldattal
- színváltás: rózsaszín-ibolya
pH≈9-10
- a kémhatást a murexid sósavas bontása után ammóniával állítjuk be
- EDTA-val titrálható a magnézium ugyanabban az oldatban
- eriokrómfekete-T indikátorral a színváltás ibolyából kékbe
vagy
- a két iont együtt mérjük pH≈9-10-es értéknél eriokrómfeketeT mellett, majd egy másik oldatrészletből a kalciumionokat 12-es pH értéknél közvetlenül titráljuk murexid jelenlétében, ekkor a két titrálás különbsége adja a magnéziumion-tartalmat
higany és cinkionok meghatározása egymás mellett
pH≈5-6
- urotropinnal állítjuk be a pH-t
- az EDTA fölöslegét visszamérjük ZnSO4 mérőoldattal metiltimolkék mellett sárgából kékig titráljuk
- Hg(SCN)42--ot választunk le az oldatból KSCN segítségével, ekkor a higanyionokkal egyenértékű EDTA szabadul föl, amit újra visszamérünk ZnSO4-tal
réz- és cinkionok meghatározása egymás mellett
pH≈9-10
- az oldatot annyi cc. ammóniával lúgosítjuk, hogy a kezdetben leváló csapadék éppen föloldódjék
- a réz- és cinkionokat így amminkomplexszé alakítva együttesen mérhetők
- murexid indikátort használva a színváltás sárgából ibolyába
pH≈5-6
- egy másik oldatrészlethez aszkorbinsavat adunk
- az elegyhez kálium-rodanidot öntünk » CuSCN leválik, a cink titrálható
- urotropin hozzáadása után a csapadékos oldatot metiltimolkék indikátort használva EDTA-val megtitráljuk
- a színváltás kékből sárgába
komplexometria nem kelatometriás módszere
cianidionok meghatározása Liebig-Denigès szerint
pH≈9
- a cianidion ezüstionnal semleges, illetve gyengén lúgos közegben stabil, vízben jól oldódó diciano-argentát komplexet képez
- 2 CN- + Ag+ = Ag(CN)2-
- a végtermék két cianidiont tartalmaz, ezért egyenértéktömege a moláris tömeg kétszerese
- az ezüstionok fölöslegével a komplex csapadékot ad:
- Ag(CN)2- + Ag+ = 2 AgCN
- ammóniás közegben azonban a végpont jelzésére jodidionokat kell alkalmaznunk, mert az ezüst-jodid nem oldódik ammóniában és nem képes megbontani a komplexet, így csak a végpont után keletkezhet:
- Ag+ + I- = AgI (sárga)
- AgNO3 mérőoldattal titrálunk
- a végpontot az oldat enyhén sárgás opálosodása jelzi
higany(II)-ionok meghatározása Volhard szerint
pH≈2
- salétromsavval állítjuk be a pH-t
- a higany(I)-tartalmat a titrálás előtt kálium-permanganáttal oxidáljuk, az oxidálószert hidrogén-peroxiddal eltávolítjuk
- KSCN mérőoldattal titrálunk, vas(III)-nitrátot használunk indikátorként
- halvány drappos rózsaszín a végpontban az oldat színe
- Hg2+ + 2 SCN- = Hg(SCN)2
- a higany egyenértéktömege a molekulatömeg fele
gravimetria
csapadékleválasztás Winkler szerint
- a csapadék leválasztása
- híg vizes oldatból
- állandó, meghatározott pH-n
- forralás közben
- cseppenként adagolt, nagy fölöslegben alkalmazott lecsapószerrel történik
- az oldathoz ammónium-kloridot adunk, ami gátolja a szennyeződések csapadékfelületre való kötődését
- a vattaszűrőt Winkler-féle kehelytölcsérben készítjük elő
- a vattaszűrő és a csapadék mosása:
- forró desztillált vízzel
- hideg desztilált vízzel
- etanollal, szívatás közben
- szárítószekrényben történő szárítás után lezárjuk a tartóedényt és tömegmérést végzünk
- a csapadék tömege az üres és a csapadékos vattaszűrő tömegének különbsége
szulfátionok meghatározása
- pH=2-es oldatból (sósavas savanyítás)
- forralás közben bárium-klorid cseppenként adagolt fölöslegével választjuk le
- Ba2+ + SO42- = BaSO4
- a csapadékot szárítószekrényben szárítjuk tömegállandóságig
- a reakcióelegyben ammónium-kloridot is oldunk
argentometria
- a pH nem lehet 8,5-nél magasabb, mert akkor Ag2O képződik
kloridionok meghatározása Mohr szerint
- semleges közegben
- kálium-kromát indikátor jelenlétében
- ezüst-nitrát mérőoldattal
- Ag+ + Cl- = AgCl
- 2 Ag+ + CrO42- = Ag2CrO4
- a Kso értékekből következik, hogy az ezüst-kromát leválása csak a kloridion-leválás ekvivalenciapontja után kezdődhet meg
- ekvivalenciapont: amikor a kálium-kromáttól sárga alapszínű oldatban a fehér csapadék enyhén drappos színűvé válik
bromidionok meghatározása adszorpciós indikátor jelenlétében
- az adszorpciós indikátor a titrálás elején adszorbeálódik az ezüstcsapadék felszínére, de a halogenidionokkal nem lép kölcsönhatásba. az egyenértékpont után a megnövekedett ezüstkoncentráció rosszul oldódó ezüstkomplexeket, s ezen keresztül színváltozást eredményez
- eozin indikátor használata esetén ecetsavval savanyítani kell az oldatot, a színváltás rózsaszínből ibolyásvörösbe
- fluoreszcein használata esetén zöldessárgából drappos rózsaszínűvé változik a csapadékos elegy
bromidionok meghatározása Volhard szerint
- a halogenidionokat ismert mennyiségű, feleslegben alkalmazott AgNO3-tal, csapadék alakjában leválasztjuk
- az ezüstionok fölöslegét lehűtött, salétromsavas oldatban vas(III)-nitrát indikátor jelenlétében KSCN mérőoldattal titráljuk maradandó halványsárgás rózsaszínig
- kloridion meghatározása esetén a csapadékos oldatot KNO3-tal forraljuk a csapadék felületének csökkentése érdekében
második féléves mérések

permanganometria
- a mérőoldat színes és egy cseppjének színe már megfelelően jelzi a végpontot
- savas közegben alkalmazzuk:
- MnO4- + 8 H+ + 5 e- = Mn2+ + 4 H2O
kálium-permanganát faktorozása
- a kálium-permanganát mérőoldatot analitikai pontossággal mért nátrium-oxaláttal faktorozzuk
- 2 MnO4- + 5 (COO)22- + 16 H+ = 2 Mn2+ + 10 CO2 + 8 H2O
- kénsavas közeget kell biztosítani, mert csak így képződik Mn2+
- a kristályos anyagot desztillált vízben oldjuk, 2N kénsavval savanyítjuk és kb. 60°C-ra melegítjük
- kristályos mangán-szulfátot adunk hozzá, mert a Mn2+ ionok katalizálják a reakciót
- kezdetben és a végpontnál cseppenként adagoljuk a permanganátot
- a végpontot az oldat egy csepp mérőoldat hozzáadására bekövetkező maradandó rózsaszínes színváltozása jelzi.
- az egyenértéktömeg az egyenlet alapján a molekulatömeg fele
peroxidok meghatározása
- kénsavas közegben a hidrogén-peroxid a permanganátionokat redukálja
- 2 MnO4- + 5 H2O2 + 6 H+ = 2 Mn2+ + 8 H2O + 5 O2
- az egyenértéktömeg a moláris tömeg fele
- így mérhető minden olyan peroxo-vegyület, amelyből savas közegben, gyors folyamatban hidrogén-peroxid képződik
- a végpontot a mérőoldat fölöslegének halvány rózsaszíne jelzi
Fe(III)-ionok meghatározása Zimmermann-Reinhardt szerint
- a vasat háromértékű formában tartalmazó, kloridion-tartalmú oldatok permanganometriás mérését teszi lehetővé
- titrálás előtt a vasat redukálni kell sósavas oldatban
- 2 FeCl4- + SnCl42- = 2 Fe2+ + SnCl62- + 6 Cl-
- kerülni kell a néhány cseppnél több ón(II)-reagens fölöslegét (forró oldatba cseppenként adagoljuk, a sárga szín eltűnése jelzi az elegendő mennyiséget)
- a redukálószer fölöslegét HgCl2-dal távolítjuk el
- ezt egyszerre adjuk hozzá, nagy feleslegben, a jól lehűtött oldathoz
- SnCl42- + 2 HgCl2 = SnCl62- + Hg2Cl2
- az oldat így kevés selyemfényű higany-I-klorid csapadékot tartalmaz
- a kloridionok zavaró hatását a Zimmermann-Reinhardt reagenssel küszöböljük ki
- a Mn(II) a kloridénál gyorsabb reakcióban Mn(III)-má oxidálódik és reakcióba lép a Fe(II)-vel
- a foszforsav a Fe(III)-mal színtelen komplexet alkot
- csökken a redoxpotenciál
- könnyebben észlelhető a végpont
- a végpontot az oldat maradandó rózsaszínes színváltozása jelzi
- az egyenértéktömeg a molekulatömeggel egyezik meg
- a módszer az összes vastartalmat méri
Bromidion meghatározása Winkler szerint
- 2 MnO4- + 10 Br- + 16 H+ = 2 Mn2+ + 8 H2O + 5 Br2
- 4 N kénsavas oldat » növeli a permanganát redoxpotenciálját
- forralás közben végezzük a titrálást
- eltávozik az illékony bróm
- csökken a bróm/bromid rendszer redoxpotenciálja
- könnyebben észlelhető a végpont
- a mérőoldatot gólyaorrú bürettából adagoljuk
- a módszer előnye, hogy a kloridionok nem zavarják
- 0,1 N permanganáttal titrálunk
- a végpontot az oldat 5-10 másodpercig állandósuló rózsaszín színváltozása jelzi
- egyenértéktömeg a moláris tömeggel egyenlő
kromatometria
- kálium-dikromát (K2CrO4) mérőoldatot használunk
- Cr2O72- + 14 H+ + 6 e- = 2 Cr3+ + 7 H2O
- előnye a permanganometriával szemben, hogy
- a mérőoldat stabilis
- nincs kloridionok által indukált reakció
- hátránya, hogy indikátor használata szükséges
- a kromatometriás titrálásokat
- kénsavval savanyított oldatban végezzük
- a végpontot difenil-amin indikátorral jelezzük
vas(II)-ionok meghatározása
- Cr2O72- + 6 Fe2+ + 14 H+ = 2 Cr3+ + 6 Fe3+ + 7 H2O
- a kloridionok jelenléte nem zavarja a reakciót
- az oldatot kénsavval kell savanyítani
- difenil-amin indikátort kell alkalmazni
- túl korán jelezné a végpontot, ezért a Fe2+/Fe3+ rendszer redoxpotenciálját lecsökkentjük a vas(III)-ionok foszforsavas komplexképzésével
- erőteljes keverés mellett, cseppenként kell titrálni, hogy a zöldesbarna átmeneti termék képződését elkerüljük
- a végpontot a halványzöld oldat határozott ibolyáskék színe jelzi
- egyenértéktömeg a moláris tömeggel egyenlő
cerimetria
- a cérium(IV)-ionok erős oxidálószerként képesek redukálódni:
- Ce4+ + e- = Ce3+
- csak erősen savas közegben léteznek
- a leggyakrabban alkalmazott indikátor a ferroin
aminofenazon meghatározása
- az aminofenazon négy elektron leadása közben oxidálható » egyenértéktömeg a moláris tömeg negyede

- aminofenazont vízben oldunk, az oldatot kénsavval savanyítjuk
- 1 csepp ferroin indikátort használunk
- végpontként azt a fogyást fogadjuk el, amelynél az indikátor zöldeskék színe pár percig megmarad
bromatometria
- kálium-bromát mérőoldatot használunk
- a meghatározások alapja lehet közvetlen oxidációs, addíciós, valamint szubsztitúciós reakció
- ezen reakciók egy része olyan gyors, hogy közvetlen meghatározásokra is alkalmassá teszi a módszert
- a többi meghatározásnál általában jodometriás visszamérést alkalmazunk
- a bromatometriás meghatározások többségét sósavas közegben kálium-bromid feleslegében végezzük
- az ekvivalenciapont jelzésére p-etoxi-krizoidin indikátort használunk
- színváltás élénk pirosból halvány sárgába
arzén(III)- vagy antimon(III)-ionok bromatometriás meghatározása
- bromát, vagy bromidionok jelenlétében az abból képződő bróm pillanatszerűen oxidálja az arzén(III)- és antimon(III)-tartalmú oldatokat:
- BrO3- + 3 H2AsO3- = 3 HAsO42- + Br- + 3 H+
- Br2 + H2AsO3- + H2O = HAsO42- + 2 Br- + 3 H+
- BrO3- + 3 SbCl4- + 6 Cl- + 6 H+ = 3 SbCl6- + Br- + 3 H2O
- Br2 + SbCl4- + 2 Cl- = SbCl6- + 2 Br-
- kálium-bromid jelenlétében sósavval savanyított oldatban 50-60°C-ra melegítve titrálunk
- indikátor: p-etoxi-krizoidin
- színváltás: élénk pirosból halvány sárgába
- 0,1 N kálium-bromát mérőoldatot használunk
- az egyenértéktömeg a moláris tömeg fele antimon esetén és a moláris tömeg negyede arzén esetén
- szerves arzén és antimonvegyületek meghatározásánál roncsolni kell azokat kénsav-peroxid eleggyel, majd redukálni kristályos hidrazínium-szulfáttal
aszkorbinsav bromatometriás meghatározása
- a reakció hideg oldatban is megfelelő sebességgel zajlik (melegítve az aszkorbinsav bomlik)

- az egyenértéktömeg a molekulatömeg fele
- az aszkorbinsavas oldatba kálium-bromidot szórunk és sósavval savanyítjuk
- p-etoxi-krizoidin jelenlétében titrálunk
- a mérőoldat 0,1 N kálium-bromát
fenazon (azofén) közvetlen bromatometriás meghatározása
- a fenazon 4-es helyzetű hidrogénje egy brómmolekulával pillanatszerűen szubsztituálódik

- azofént vízben oldunk és sósavat adunk hozzá
- az oldatban kálium-bromidot oldunk
- p-etoxi-krizoidin indikátort adunk hozzá
- állandó keverés mellett lassan titrálunk
- a 0,1 N kálium-bromát mérőoldatot addig adagoljuk, amíg a piros szín sárgára nem változik
- a túltitrálás elkerülése érdekében kellő KBr-fölöslegre van szükség
- egyenértéktömeg a moláris tömeg fele
Jodometria
~0.01 N Na-tioszulfát mérőoldat faktorozása
- pontos beméréssel nem lehet készíteni, ezért 1.000 faktorú KIO3 vagy kálium-hidrogén-jodát mérőoldattal ellenőrizzük
- gyengén savanyú oldatban (pH<5) kellő sebességgel megy végbe
- az ismert mennyiségű KIO3 vagy KH(IO3)2 mérőoldatot vízzel hígítjuk, sósavval savanyítjuk
- az oldatba kálium-jodidot szórunk
- IO3- + 5 I- + 6 H+ = 3 I2 + 3 H2O
- a kivált jódot titráljuk a faktorozandó Na-tioszulfáttal
- végpont előtt keményítő indikátort adunk hozzá
- a kék szín eltűnéséig titráljuk (1-2 perc elteltével visszakékül)
- az egyenértéktömeg a molekulatömeg hatoda jodát esetén és Mt/12 hidrogén-bijodát esetén
Fenol meghatározása Koppeschaar szerint
- lépcsőzetes brómszubsztitúció, a hosszú reakcióidő miatt visszaméréses titrálást alkalmazunk
- ismert bromát mérőoldat-felesleget kell alkalmaznunk és az el nem reagált brómot határozzuk meg jodometriásan
- a fenolt a kálium-bromát, a kálium-bromid és a sósav reakciójában szabaddá váló bróm feleslegével brómozzuk
- BrO3- + 5 Br- + 6 H+ = 3 Br2 + 3 H2O

- a bróm fölöslegét jodometriásan visszamérjük:
- kálium-jodidot adunk hozzá
- Br2 + 2 I- = 2 Br- + I2
- kivált jódot 0.01 N Na-tioszulfáttal titráljuk meg
- I2 + 2 S2O32- = 2 I- + S4O62-
- végpont közelében keményítő indikátort adunk hozzá
- Egyenértéktömeg: Mt/6
- színváltozás: jódkeményítő kék színének eltűnéséig
acetilszalicilsav-tartalom meghatározása tablettákban
- az acetil-szalicilsav lúgos hidrolízise során acetát- és szalicilátionok képződnek

- átsavanyítás után a megfelelő savakká alakulnak
- a szalicilsav brómfelesleg hatására tribróm fenollá alakul

- ugyanúgy mérhető, mint a fenol (Koppeschaar)
- a tabletta porított részletét 2M NaOH és víz elegyében oldjuk
- 10 perces forralás után hűtjük és törzsoldatot készítünk
- kálium-bromát fölöslegét és kálium-bromidot adunk hozzá
- sósavval savanyítjuk és egy órát állni hagyjuk
- a bróm fölöslegét Koppeschaar szerint mérjük, 0,1 N Na2S2O3 mérőoldattal (visszamérés)
- az egyenértéktömeg a molekulatömeg hatoda
Na-hipoklorit-tartalom meghatározása
- hipohalogenitek savas közegben KI feleslegével reagálva ekvivalens mennyiségű jódot választanak ki:
- ClO- + 2 H+ + 2 I- = Cl- + I2 + H2O
- a gyenge savas közegben a diszproporcionálódással képződő klorát csak lassan oxidálja a jódot:
- 3 ClO- = ClO3- + 2 Cl-
- vízben kálium-jodidot oldunk és sósavval savanyítjuk, ebbe pipettázzuk a hipoklorit tartalmú oldat részletét
- 0,01 N Na-tioszulfát mérőoldattal titráljuk
- végpont közelében keményítő indikátort adunk hozzá
- színváltozás: jód kék színének eltűnése
- egyenértéktömeg: a molekulatömeg fele
króm(III) (kromát- és dikromátionok) meghatározása
- króm(III)-ion lúgos oldatban hidrogén-peroxiddal forralva kvantitatívan kromáttá oxidálható:
- 2 Cr(OH)4- + 3 H2O2 + 2 OH- = 2 CrO42- + 8 H2O
- hidrogén-peroxid feleslegét elforraljuk (ezt az ezüstionok katalizálják)
- 2 H2O2 = O2 + 2 H2O
- a krómion így meghatározhatóvá válik
- kromátionok és a savanyításra belőlük képződő dikromátionok erősen savas közegben a jodidionokat jóddá oxidálják
- Cr2O72- + 6 I- + 14 H+ = 2 Cr3+ + 3 I2 + 7 H2O
- az erősen savas közeg miatt a levegő O2-jének zavaró hatását kell kiküszöbölni (jodid → jód oxidáció)
- KHCO3-ot szórunk az oldathoz a kálium jodid hozzáadása előtt
- a fejlődő CO2 kiszorítja az oxigént a lombikból
- a mérés során a Cr(III)-ionokat tartalmazó oldat részletét vízzel kiegészítve NaOH-dal és cc. H2O2-dal elegyítjük
- kis lánggal forraljuk, ezüst-nitrátot juttatunk az oldatba (peroxidbontás)
- szobahőmérsékletűre hűtjük és kénsavval savanyítjuk
- kálium-hidrogén-karbonátot szórunk hozzá
- a pezsgés megszűnésekor hozzáadjuk a kálium-jodidot
- a kivált jódot 0,01 N Na-tioszulfát mérőoldattal titráljuk
- a keményítő indikátort csak a végpont előtt adjuk hozzá
- a végpont a króm(III)ionoktól halványzöld oldat
- egyenértéktömeg a molekulatömeg hatoda dikromát esetén, króm(III) esetén a molekulatömeg harmada
KI meghatározása Winkler szerint (sokszorozó módszer)
- gyengén savanyú oldatban végezzük a titrálást
- sósavval savanyítunk
- pH=2-n kvantitatív a jód oxidációja
- a klór diszproporcionálódása el sem kezdődik
- klóros víz feleslegével jodáttá oxidáljuk a jodidot (annyi klóros vizet adagolunk, hogy a kivált jód elszíntelenítéséhez fölhasznált mennyiségnek még másfélszeresét vesszük)
- I- + 3 Cl2 + 3 H2O = IO3- + 6 Cl- + 6 H+
- a klórfelesleget kiforraljuk
- akkor forraltuk ki teljesen a klórt, ha a hozzácseppentett metilnarancs megtartja színét
- az oldatot forralás után lehűtjük és kálium-jodidot oldunk benne (redukálódik a jodát)
- IO3- + 5 I- + 6 H+ = 3 I2 + 3 H2O
- foszforsavval savanyítjuk és a kivált jódot tioszulfát mérőoldattal titráljuk:
- 3 I2 + 6 S2O32- = 6 I- + 3 S4O62-
- keményítőt csepegtetünk bele a végpont előtt
- a végpontban visszakapjuk a metilnarancsos színt
- egyenértéktömeg a moláris tömeg hatod része
- klorid és bromid nem zavarja az eljárást (kiforralás miatt)
- az eljárás alkalmas jódozott konyhasó jódtartalmának meghatározására
Réz(II)ion jodometriás meghatározása
- 2 Cu2+ + 4 I- = 2 CuI + I2
- réz(I)-jodid kis oldhatósága teszi lehetővé
- a réz(II)/réz(I) rendszer katalizálja jodid oxidációját
- ecetsavval savanyítunk, hogy csökkentsük a hatást
- a vizsgálandó oldat részletéhez vizet öntünk, ecetsavval savanyítjuk
- az elegyben kálium-jodidot oldunk
- 20 perc elteltével a kivált jódot 0,01 N Na-tioszulfáttal titráljuk
- keményítő indikátor a végpont előtt
- végpont: legalább 1 percig színtelen - fehér - marad
- egyenértéktömeg a molekulatömeggel egyezik meg
Mannit meghatározása (Malaprade reakció)
- polialkoholok esetén mindig 2 mol formaldehid képződik, a keletkező hangyasav mólszáma pedig a szekunder alkoholcsoportok számával egyezik

- a Malaprade-reakció során fölöslegben maradt perjodátot nem határozhatjuk meg egyszerűen jodometriásan:
- IO4- + 7 I- + 8 H+ = 4 I2 + 4 H2O
- mivel az alkohol reakciója során képződő jodát is jódképzés közben reagálna
- ha pH>5 (NaHCO3-dal tompított oldatban pH~8) a jodát már nem képes oxidálni a jodidionokat, csak a perjodát reagál:
- IO4- + 2 I- + 2 H+ = I2 + IO3- + H2O
- a kivált jódot nátrium-arzenit mérőoldattal kell titrálnunk (ezen a pH-n a jód-tioszulfát reakció nem egyértelmű)
- I2 + AsO33- + H2O = 2 I- + AsO43- + 2 H+
- az egyenértéktömeg a molekulatömeg tizede
- a mérés során a törzsoldat gyengén ecetsavas részletét perjódsavoldattal elegyítjük, hígítjuk
- Na-hidrogén-karbonátot szórunk bele apró részletekben
- kálium-jodidot szórunk az oldatba
- a kivált jódot 0,1 N Na-arzenittel titráljuk keményítő jelenlétében
- a méréssel párhuzamosan üres mérést végzünk csak a kémszerekkel, a fogyást a két mérés különbségéből kapjuk
műszeres mérések
tojáshéj foszfortartalmának spektrofotometriás meghatározása
- a molibdátion a foszfáttal dodekamolibdáto-foszfát-ionok képződése közben reagál:
- H2PO4- + 12 HMoO4- + 10 H+ = PMo12O403- + 12 H2O
- a képződött komplex aszkorbinsavval szelektíven kék, molibdén(V)-öt tartalmazó termékké redukálható:
- PMo12O403- + n e- = PMonVMoVI12-nO40-3-n
- a kék anyag mennyisége spektrofotometriásan mérhető
- a tojások héját mossuk, szárítjuk, majd hamvasztjuk
- analitikai pontossággal bemért mennyiségét 1:2 hígítású sósavban oldjuk fel, ebből készítjük a törzsoldatot
- kalibráló oldatsorozatot készítünk, az előkészített oldatokhoz kénsavat, ammónium-molibdátot és aszkorbinsavat adunk
- a szín kifejlődése időreakció, állás után mérjük az oldatok abszorbanciáját adott hullámhosszon
- az abszorbanciaértékeket a koncentráció függvényében ábrázolva a koncentrációkat grafikusan határozhatjuk meg
Elixirium thymi compositum bromidtartalmának mérése potenciometriásan
- a készítmény sötétbarna színű, így az argentometria minden vizuális végpontjelzését lehetetlenné teszi
- bromidtartalma potenciometriásan határozható meg
- NaOH-dal semlegesítjük, ecetsav-acetát pufferral állítjuk be a pH-t
- ezüst mérőelektródot és kettős sóhidas másodfajú (Ag/AgCl/KCl/KNO3) referenciaelektródot használunk
- ezüst-nitrát mérőoldattal titrálunk
fogkrém fluorid-tartalmának meghatározása potenciometriásan
- a méréseket fluoridion-szelektív elektróddal végezzük, az érzékelő lantán-fluorid, amelynek vezetőképességét európium(III)-mal növelték
- a fluoridion aktivitását állandó értéken kell tartanunk, ezért TISAB-oldatot használunk
- az ionerősséget és a pH-t is szabályozza
- nátrium-klorid, ecetsav, nátrium-acetát
- konstans ionerősséget biztosít
- ecetsav és az acetát
- pufferolja a pH-t
- nátrium-citrát
- komplexképzőként a fluorid mérését zavaró fémnyomokat (Fe(III)-ion) maszkírozza
- referenciaelektródként KCl-dal töltött Ag/AgCl elektródot (KNO3 sóhíddal) használunk
- kalibrálósorozatot készítünk, ábrázoljuk az EME értékeket a lg cF- függvényében
- a meghatározandó koncentrációkat erről a grafikonról leolvasott értékek alapján számoljuk
foszforsav mérése NaOH-dal cola-ban, potenciometriás titrálás számítógépvezérléssel
- a számítógép megfelelő interface-ek segítségével az analitikai mérőműszerek irányítására és szabályozására is alkalmas
- az automata potenciometriás titráló berendezéshez digitális pH-mérőt és automata bürettát használunk
- mérés közben a bevitt térfogat függvényében rögzítjük az egyensúlyi potenciált, illetve a pH-t
- a foszforsav háromértékű sav, a savi disszociációs állandók közötti megfelelő különbség lehetővé teszi, hogy több lépcsőben titráljuk NaOH-dal
- a pH-mérőt két pufferre kalibráljuk
- a cola-ból eltávolítjuk a CO2-ot, és belehelyezzük a kombinált üvegelektródot
- nincs lehetőség a végpont előtti kiforralásra, ezért karbonátmentes lúg mérőoldatot kell használni, ezt a légszívó csonk bemeneténél lévő Ba(OH)2-os mosóval érhetjük el
nagyhatékonyságú folyadékkromatográfiás analízis (HPLC)
- széleskörű alkalmazhatóságát számos előnyös tulajdonsága teszi lehetővé
- az analízis szobahőmérsékleten történik, így hőérzékeny vegyületek, biológiai minták is vizsgálhatók
- legfőbb hátránya magas költsége
- HPLC készülék fölépítése:
- oldószer tároló edény » oldószer keverő » szűrő » pumpa » nyomás- és áramlásmérő » injektor » előtétkolonna » analitikai kolonna » detektor » adatrögzítő és -értékelő egység
- valamennyi detektor a vizsgált rendszer valamely fizikai vagy kémiai paraméterével egyértelmű függvénykapcsolatban álló jelet ad
- ha több komponenst vizsgálunk, a detektornak alkalmasnak kell lennie valamennyi kimutatására
- leggyakrabban UV-VIS fotometriás detektorokat használnak, bár ezek alkalmazásának feltétele a kromofór csoport
- a leggyakrabban használt kolonnák szilícium-dioxid szemcséket tartalmaznak, melyek mérete eltérő
- a poláris, kissé savas szemcsék felületét módosíthatják, ezzel apolárissá téve azokat
- elegyek komponenseinek elválasztásakor az tekinthető optimálisnak, ha az egyes komponensek teljes egészében elkülönülnek, a vizsgálat a lehető legrövidebb ideig tart, és a csúcsok alakja is elfogadható
- k, kapacitási tényező egyetlen csúcs jellemzésére használható
- k, = (tR - t0) / t0
- két csúcs elkülönülését a csúcsfelbontással jellemezhetjük
- RS = (tR2 - tR1) / (W1 + W2)
- ahol tR a retenciós idő és W a csúcs félértékszélessége
ionkromatográfia
- az ioncserekromatográfia és a HPLC kombinációja
- 2 ioncserélő oszlop (szeparátor és szupresszor)
- nagy nyomás
- vezetőképességi detektálás
- a szeparátor anion- (R-N+⋅⋅⋅HCO3-) vagy kationcserélő oszlop, a szupresszor az eluens vezetőképességének elnyomására szolgál (R-SO3-⋅⋅⋅H+)
- anionok sorrendje: F-, Cl-, Br-, NO3-, H2PO4-, SO42-
- a tengelyeken: μS/cm vs. min
- a szeparátor anioncserélő regenerálása NaHCO3 eluenssel
- a szupresszor kationcserélő regenerálása kénsavval, vízzel
félkvantitatív iontesztek
- szelektív és érzékeny színreakciók alapján gyors, félkvantitatív eredmény érhető el
- a reakcióidő max. 2 perc
- a reakciózónán jellegzetes színváltozás lép föl, amely a színskálákkal összehasonlítva lehetővé teszi a koncentráció meghatározását
többértékű gyenge savak mérése vörösborban konduktometriásan
- a borokban lévő savak gyenge savként titrálhatók lúg mérőoldattal
- a bor színét adó anyagok sav-bázis indikátorként működnek, azonban nem a titrálás végpontjában váltanak színt, ezért a vizuális végpontjelzést lehetetlenné teszik
- a gyenge savak erős bázissal titrálva nem határozhatók meg konduktometriásan, mert nem a hidrogénion-koncentráció csökkenése, hanem a keletkező ionok koncentrációjának növekedése okozza az oldat vezetőképességének változását
- ha nem erős, hanem gyenge bázist használunk mérőoldatként, az ekvivalenciapont után a vezetőképesség már nem változik
- vezetőképességi elektródot (harangelektród) és NH3 mérőoldatot használunk
atomabszorpciós spektroszkópia
- alapállapotú atomok képesek megfelelő hullámhosszúságú fény elnyelésére, ha az atomgőzt a jelenlévő elemek színképvonalához tartozó hullámhosszúságú fénnyel sugározzuk be, a vonal helyén éles abszorpció történik
- atomgőz létrehozására két módszer használatos:
- lángtechnika:
- a mintát a lángba porlasztjuk, a jel folyamatos, a kiértékelés a csúcsmagasság alapján történik
- jelentős mértékű a mintafelhasználás, de kicsi a mérések szórása, az atomok a fényútban rövid ideig tartózkodnak, ezért a kimutatási határ ppm tartományban van
- grafitkemencés módszer:
- a minta egy grafitból készült küvettából párolog el, a jel tranziens és a kiértékelés a csúcs alatti terület alapján történik
- a mintafelhasználás kicsi, a mérések közötti szórás nagyobb, a minta hosszabb időt tölt a fényútban, így ppb tartományban van a kimutatási határ
- az atomgőzön állandó intenzitású, monokromatikus fénysugarat bocsátunk át
- a nagy felbontású monokromátorokat, vagy vájtkatód lámpákat használunk
- a vájtkatód lámpában az anód volfrám, a katód ameghatározandó fém, kis nyomású nemesgázzal van töltve
- feszültséget kapcsolva a lámpára a kisülés porlasztja a katódot, majd gerjeszti az elektronokat, amelyek relaxációjukkor a rájuk jellemző spektrumot sugározzák
- a vájtkatód lámpákhoz nem kell monokromátort használni
- a láng hőmérsékletét az éghető és égést tápláló gáz mennyiségének megválasztásával szabályozhatjuk
- a detektor régebben fotocella, ma általánosan fotoelektronsokszorozó
- a mérés:
- kikeressük a kalcium méréséhez szükséges műszerparamétereket
- a meghatározáshoz kalibráló oldatsorozatot készítünk
- a zavaró hatások kiküszöbölésével is foglalkoznunk kell
- ellenőrizzük, hogy a kalcium vonalán megfelelő intenzitás jut-e a detektorba
- begyújtjuk a lángot
- injektáljuk a legmagasabb koncentrációjú standardot
- beállítjuk a lámpát, a lámpaáramot és kiválasztjuk a megfelelő hullámhosszt
- megvizsgáljuk, hogy a kiválasztott hullámhossz értéken kapjuk-e a legmagasabb transzmittanciát
- a lámpaáram egy bizonyos szintig javítja a jelet, utána azonban csak a lámpa élettartamát csökkenti
- beállítjuk a résszélességet és a láng pozícióját (magasság, forgatással az úthossz csökkentése)
- végül a láng összetételét és a porlasztás sebességét változtatjuk
- az abszorbancia értékeket ábrázoljuk a koncentráció függvényében, a kalibráló görbe segítségével értékeljük ki a mérést